This epitope scanning tool aims to identify radical amino acid substitutions (mutations), in this case in the Omicron variant of the SARS-CoV-2 virus, and to determine if such mutations hit relevant epitopes (B-cell, T-cell class I and II). The data was obtained from Global Initiative on Sharing All Influenza Data (GISAID) at www.gisaid.org.

Explanation
Epitopes are like unique identifiers/attributes of a virus (SARS-CoV-2) that the body remembers either through natural infection of through vaccines such that the body will recognize a similar virus that enters in the future. The idea is that if several of those attributes are radically mutated, then the body will have difficulty identifying the virus based on the attributes of the former virus.
What are the locations of the epitopes?
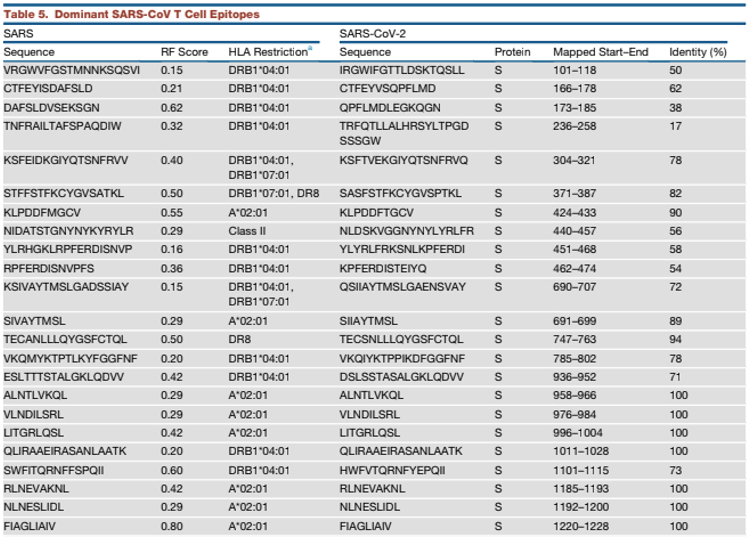
Grifoni et. al. [1] identified 28 epitopes (5 B-Cell and 23 T-Cell) in the Spike protein region of SARS-CoV-2 based on its similarities ("Identity %" column) with SARS-CoV. See tables below for their respective locations ("Mapped Start-End" column) in the Spike protein region:
What are the locations of the epitopes?
Grifoni et. al. [1] identified 28 epitopes (5 B-Cell and 23 T-Cell) in the Spike protein region of SARS-CoV-2 based on its similarities ("Identity %" column) with SARS-CoV. See tables below for their respective locations ("Mapped Start-End" column) in the Spike protein region:
Which mutations are potentially harmful?
Grantham [2] index was used to classify the mutations into either conservative or radical mutations. Note that we used the Wuhan variant of SARS-CoV-2 as the baseline or reference genome from which to compare the amino acid substitutions/changes/mutations. Radical mutations in the epitope regions of the Omicron variant of SARS-CoV-2 have considerable impacts in the transmissibility and disease severity of the virus. The Grantham index D is given by the following distance formula:
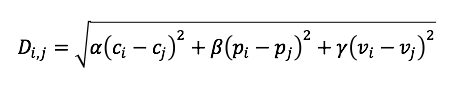
where c, p, and v are the composition, polarity, and molecular volume of the amino acid; and alpha, beta, and gamma are scaling constants. A mutation with Grantham index D >100 is classified as a radical mutation [3]. The table below shows the Grantham values of the mutations to-and-from the 20 known amino acids (denoted by their corresponding single character symbols):
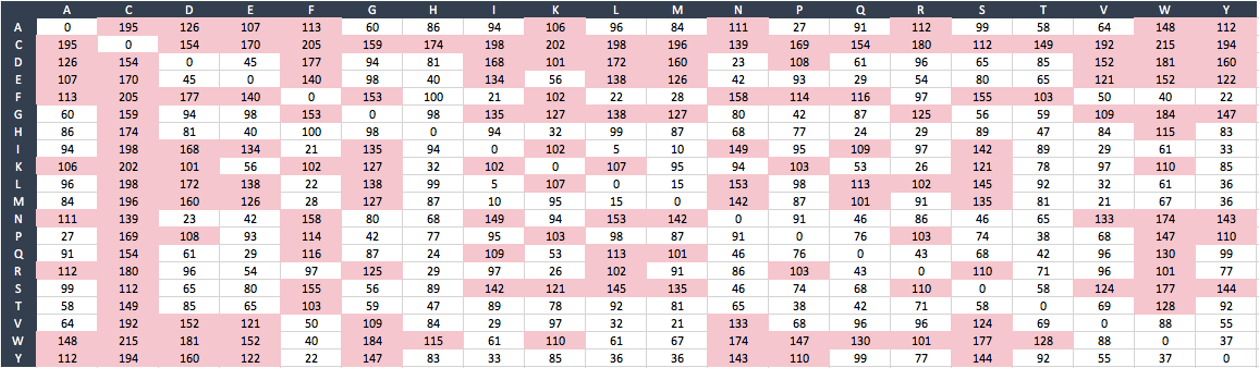
For example, when amino acid Alanine (denoted by "A") is replaced by amino acid Cysteine (denoted by "C") or vice versa, such change or mutation is classified as radical given by the Grantham index = 195.
How to interpret the epitope scanning tool?
One way to use the epitope scanning tool is to count the number of radical mutations that hit the epitopes. Then count the number of samples that exhibited such behavior. In this case, we used the first 200 sequenced samples of Omicron variant from the GISAID database. The assumption is that when the proportion of the epitopes are hit by radical mutations is high (>50%), then this variant is radically different from the reference genome (e.g. Wuhan variant).
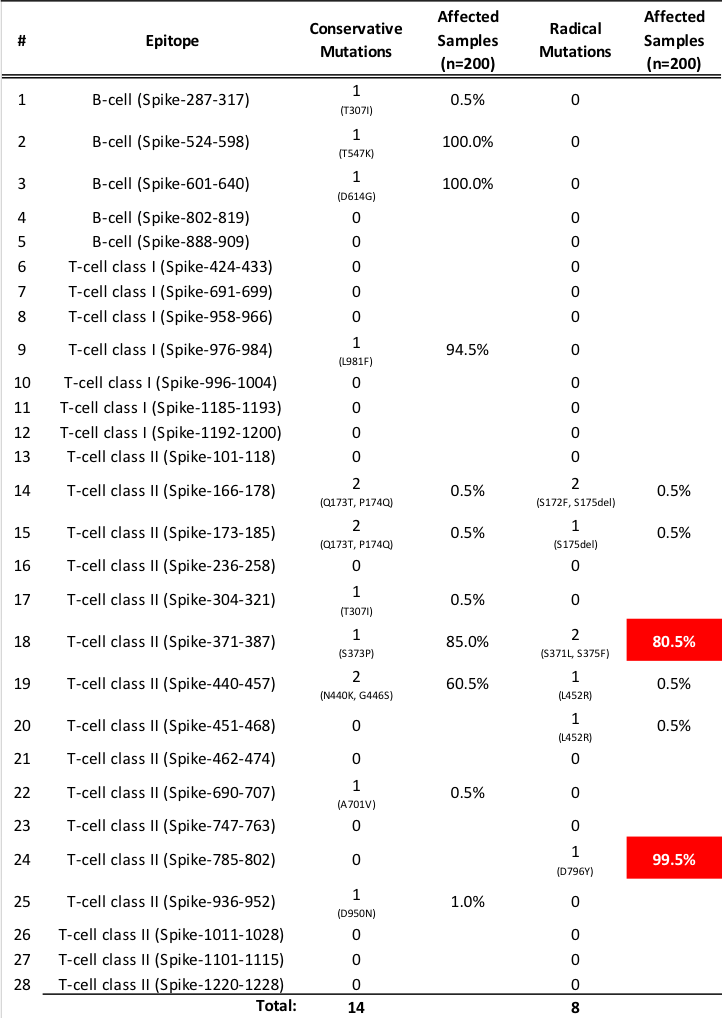
Looking at the summary table above, the Omicron variant may have increased transmissibility and disease severity due to several mutations hitting the T-Cell epitope (class II). But only 2 out of the 28 epitopes were convincingly hit by radical mutations (S371L, S375F, D796Y). Thus, a human body’s immune system may still be able to recognize the remaining 26 epitopes in Spike protein with the help of the COVID-19 vaccines that are based on the Wuhan variant.
[1] Grifoni et al., 2020, Cell Host & Microbe 27, 671–680. April 8, 2020 a 2020 Elsevier Inc. https://doi.org/10.1016/j.chom.2020.03.002
[2] Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974 Sep 6;185(4154):862-4. doi: 10.1126/science.185.4154.862. PMID: 4843792.
[3] Li W-H, Wu C-I & Luo C-C (1985) A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Molecular Biology and Evolution 2:150-174.
[2] Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974 Sep 6;185(4154):862-4. doi: 10.1126/science.185.4154.862. PMID: 4843792.
[3] Li W-H, Wu C-I & Luo C-C (1985) A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Molecular Biology and Evolution 2:150-174.